BEAP ASSEMBLY TOOL
USER GUIDE
Software developed by Zhiliang Hu, Eric Fritz
Documentation developed by James Koltes, Eric Fritz, and Zhiliang Hu
July 14, 2008
Overview
BEAP, the BLAST Extension and Alignment Tool, was developed as a method to expedite the assembly of localized genomic regions. The amount of sequence capable of being assembled by BEAP varies greatly depending on computation power and number of matching sequences available in the target database. In general, 1Mb is a maximum amount that the tool can assemble in the given situation tested (see examples in the paper), however larger assemblies (less than 10Mb) may be possible with sufficient computing power. BEAP is not designed as a genome assembly tool, rather, Its intended use is to assist researchers working with genomes that have limited or incomplete genomic sequence requiring assembly in a small genomic region. The viewer GUI is added to allow users visually assess the quality and SNP content of a given set of assembled sequences within a contig. We have tried to make the tool configurable so that it may be adapted for more versatile use.
The BEAP Tool
BEAP is a unix command-line program that employs NCBI blast (MegaBLAST or netBLAST) and CAP3 assembly tools to "blast and extend" sequences based on available similar sequences it can find from given target databases.
The BEAP assembly tool facilitates sequence assembly using a few "priming" sequences, usually short sequences such as EST, mRNA, etc., to blast against a number of NCBI sequence databases in a cycling fashion, i.e. matched sequences from the first blast round are used as new "query" sequences in the 2nd round blast, and so on. The result is a local sequence database containing similar sequences for use in assembly. The number of BLAST rounds performed can vary based on need. Any DNA sequence that was retrieved multiple times was removed. Sequences were then assembled into contigs by CAP3. Once an output file is saved, user may upload the resulting .ace into the BEAP GUI, where sequences can be viewed in either a contig or sequence view (details provided below.) Using the GUI, contig sequence quality, and predicted SNP content can be determined. Full length contig sequence as well as individual sequence members can be easily accessed for further use.
Input: A "priming" sequence in fasta format.
Output: All sequences found by recursive blast matches "assembled" in CAP3 alignments, along with alignment quality scores, among other standard CAP3 outputs.
Steps to run the program 
If you are ready to try it out, we assume all pre-requests are satisfied as specified in the README, namely: (a) A local copy of NCBI Blast Suite (version 2.2.23) is installed; (b) Target blast sequence database are prepared (see Blast manual for details). (c) A local copy of cap3 is installed; (c) perl (version 5.8 or later) are installed.
- Unpackage the downloaded tar ball:
> tar zxf beap_v.xxx.conf.tar.gz
This will expand the download into 4 files:beap_v.0.6 beap_v.0.6.conf BeapViewer5.3.jar README.txt
Check the first line in "beap_v.0.6" to make sure it points to the correct perl installation on your computer. - Prep run: Simply run the program with a command line call:
> beap
For the first time you run it, it will generate a user-configurable "beap.conf" file for you (Alternatively, you can manually copy the configuratoin template, "beap_v.xxx.conf" to "beap.conf", to be modified and used for your BEAP run).Use a unix editor such as 'vi' or 'pico' to open and change the values for each parameter in the "beap.conf" file to fit your computer environment (the template "beap.conf" file is self-explanary; See "Configurations" section below for more details).
- Input sequence prep: Prepare an input sequence file in fasta format, with the file name as you specified in your configuration file.
- Run the program: As it may take substential amount of time for
a run, it's recommanded to send your job to run in the background, as in:
> beap &
The output will be saved into several files, with the file names pre-fixed with a "root name" as you specify in your configuration file.
Configurations
The input file, output file, blast engine to use, target blast databases, and blast strengencies among other options are stored in a configuration file called: "beap.conf", located in your working directory. (Hint: you can run multiple jobs, as long as you keep all files in a run in its own directory. For example, it would be a good preactice to create new directories for each trial you wish to run.
Once the configuration parameters are set with proper values, you can start the run, by typing the command "beap" as shown above. Since this process may take quite some time, from within an hour to several hours depending on your job, you may want to send the process to the background to avoid being interrupted.
If your computer has a stable internet connection, "netBlast" can give you much flexibility and power by allowing you to utilize NCBI blast facility (see Appendix A, B, C in the netBlast menual for target databases among other options to use in the BEAP configuration file).
More on configuration -- optimize the options
We tested different settings to determine how to maximize contig number and size produced by BEAP in normal situations. Using network BLAST resulted in most sequence queried and contigs built. However, sometimes, computational time for network BLAST required hours compared to minutes for local BLAST depending on network availability. In addition, contig sizes were generally the same for network versus megaBLAST. It was difficult to assess the difference in terms of efficiency and productivity of the two. This would suggest that megaBLAST on a local database is considerably faster for creating contigs with BEAP. As expected, increasing the E-value to e-30 compared to e-60 or e-120 resulted in the most sequence queried and contigs created. Changing the word size in megaBLAST likewise resulted in the most contigs when the word size was smallest, such as 10, versus setting of 30 or 60. The idea of changing word size is to require a larger string of nucleotide matches for BLAST to classify a sequence as a good match to the template. However, there was very little difference in the number or size of contigs created using a word size ranging from 10 - 30. Users should realize that using local BLAST requires frequent sequence downloads to access the most up-to-date sequence resources. NetBLAST has the advantage of always accessing the most up-to-date list of sequences available at NCBI.
Based on these preliminary trials, the optimal setting to produce contigs with BEAP may be recommended as follows. The user should download as many databases as possible, updating them as often as possible, onto a local server with considerable computational power (8-16 Gb RAM). One way to form a template sequence isby comparing the species of interest to a fully sequenced "template" genome using a RH map. The "template" sequence would be composed of 1 Mbs of sequence filtered for repetitive sequence with a program like Repeat Masker (http://www.repeatmasker.org/). Then, the megaBLAST option could be used to query the desired local databases with an E-value of e-30 and a word size of 10 or smaller. Lower E-values or smaller chunks of template sequence could be used to compensate for large template input sequence sizes greater than 1Mbs. The E-value setting could be adjusted higher or lower to retrieve more or less sequence for the desired specificity and sensitivity or retrieved sequence required of the user.
Technical Specs
(See each Release for details).
The BEAP Extension: A GUI Viewer
The graphical GUI is programmed in java. It's designed to display the sequence alignments from the BEAP program. Once the resulting sequences from cap3 is ready,they can be imported into the BEAP Viewerto assess the sequence alignment, quality and presence of possible SNPs.
Input: The input file is from BEAP (cap3) output, in .ace file format. An input file can be opened with the Upload .ace file button.
Views: The sequence alignments can be viewed in two ways:
- Line overlap representation view, and
- Sequence alignment view.
The view of the two modes can be switched by choosing respective tag from the View pull-down menu. Only one contig alignment can be viewed at a time. When there are multiple contigs in one .ace file, use the Choose the Contig to View pull-down menu to select the contig to view.
Link out: More detailed information on the alignment sequences can be browsed to with built-in link functions. Click on a sequence name or a line to open up a new window with the complete sequence of that member.
Output:
- Graphic representation of the alignment may be saved into a .jpg file for records.
- The alignment may also be printed.
Description of GUI Use
Once you have uploaded a file and a contig is displayed (Figure 1A), there are different options available.
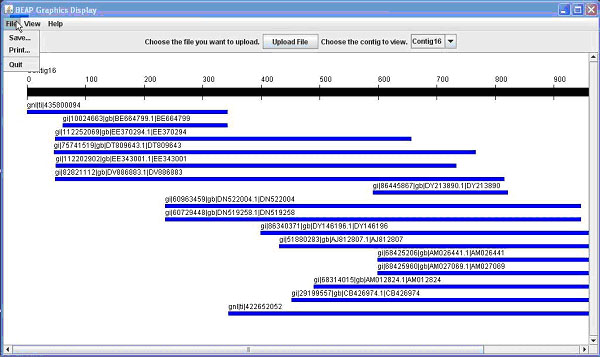
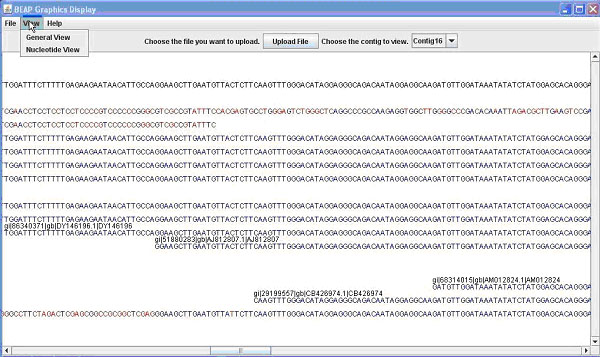
Figure 1. The BEAP G.U.I. This comparison shows both 1A (above) the General View; and 1B (below) the Nucleotide View.
The first option is to save the view of the contig; this would be useful for presentations or publications. A second option available is to print the view, which allows for writing notes or marking important areas on the contig. Another option would be to click on an individual line representing a sequence to open another window shown in Figure 2.
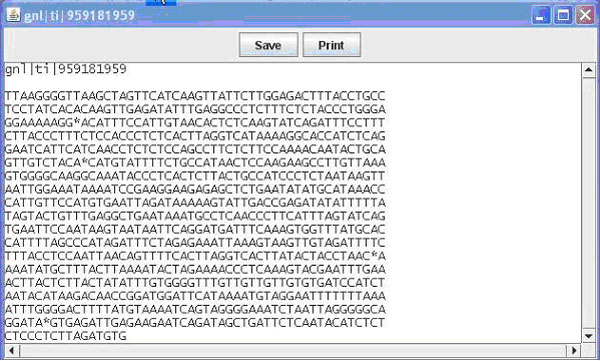
Figure 2. The pop up window displaying an individual sequence. This window is available upon clicking one of the sequences in the main window when viewing the General View of the contig.
Contained in this window is the name (e.g. EST name, Bac name) of the sequence as well as the sequence. From this window you can once again save or print what is displayed. Finally from the main window you can switch the view to nucleotide view, which displays the contig and sequences as before, this time display the individual bases that make up the sequence (Figure 1B). Bases in a sequence that do not match with the contig are marked in red for ease of finding. This allows the user to easily identify portions of sequence that are inadequate and to quickly find areas of possible single nucleotide polymorphisms (SNPs). Additional details on use of the GUI can be obtained in the BEAP viewer manual.