version 1.0 (June 2008)
The MIQAS_TAB set of file formats are designed to describe the results from a QTL or association study according to MIQAS minimal requirements. These files are all tab-delimited and provide an alternative solution for data exchange from MIQAS_XML, the XMLSchema provided with the MIQAS standard. Even though the information described here could be entered in different sheets of a spreadsheet application like Microsoft Excel or OpenOffice, the MIQAS_TAB specification explicitly refers to tab-delimited text files. One of the main reasons is the fact that spreadsheet programs can alter data in unexpected ways.
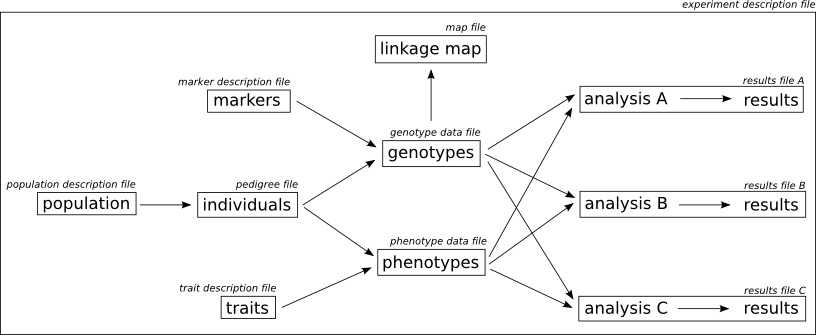
The different sections of the MIQAS standard are covered by one or more tab-delimited files. The main file, called experiment_description_file.txt, provides meta-data for the study and links the rest of the data into a coherent whole. When one submits data using MIQAS_TAB, the researcher sends a group of files and indicates which of these is the experiment description file. A system loading this data will use the information in that file to locate the rest of the data. An overview of the conceptual parts covered by the MIQAS_TAB files is presented in Figure 1.
The following files are used:
Some of these files are always required, while others are optional under certain circumstances. See the documentation for the experiment description file (below) for more information.
There are two alternative formats for these files: either they contain two columns (i.e. tag-value pairs) or are a grid of data.
The id field mentioned in several of the files acts as an internal reference within the group of files. No additional meaning should be attributed to it.
The experiment description file contains meta-data for the study and links to the rest of the data. Rows in this file normally contain 2 columns (with one exception), following a tag-value paradigm: the first column provides a tag while the second column is the value for that tag.
Data provided in this file:
At minimum, the specification version, a title and a contact email have to be provided, as well as names for some of the other files. Entries for related result contain an extra column indicating the type of relationship.
Files that must be provided are the experiment description file, the population description file, the trait file and the marker file. Some files are mandatory under certain circumstances: the qtl file and map file are mandatory if the experiment describes a QTL study. The association file is mandatory if the experiment describes an association study. In addition, the pedigree file is mandatory if either the genotype or phenotype file is present.
Typical issues with this file:
| specification version | 1.0 | |
| title | Roslin Institute QTL study 123 | |
| description | this is what the study is about | |
| contact last name | Aerts | |
| contact first name | Jan | |
| contact email | jan.aerts@myisp.com | |
| reference PMID | 12345678 | |
| related result | ArkDB|ARKQTL00000001 | fine_mapping |
| population description file | population.txt | |
| pedigree file | pedigree.txt | |
| trait description file | traits.txt | |
| marker description file | markers.txt | |
| genotype data file | genotypes.txt | |
| phenotype data file | phenotypes.txt | |
| map file | map.txt | |
| results file | results_first_analysis.txt | |
| results file | results_second_analysis.txt |
The population description file contains information of the population. Like the experiment description file, the population description file contains two columns with tag-value pairs.
Data provided in this file:
| species | Bos taurus |
| description | BovGen – cross between extreme phenotypes for response for bacterial infection |
| source | Holstein x Belgian Blue |
| structure | intercross |
The pedigree file contains information on the sex of the individuals and the relationships between them. This file is grid-based. The four columns contain the individual id (as used in the genotype and phenotype data files), the sex, the id of the father and the id of the mother. Only the first column is mandatory. The first row contains the headers: ”#individual_id”, “sex”, “father_id” and “mother_id”. Values for sex can be “UNKNOWN”, “MALE”, “FEMALE” or “MALE&FEMALE” (e.g. for oyster). Unknown values should be reported as “UNKNOWN”. Both the father_id and mother_id must be values that are also present in the individual_id column.
This file is mandatory if genotypes or phenotypes are provided (even if it just would contain a list of IDs). If neither the genotypes and phenotypes file is given, the pedigree file is still recommended.
Typical issues with this file:
| #individual_id | sex | father_id | mother_id |
| ind_1 | MALE | UNKNOWN | UNKNOWN |
| ind_2 | FEMALE | UNKNOWN | UNKNOWN |
| ind_3 | MALE | ind_1 | ind_2 |
| ind_4 | MALE | ind_3 | ind_2 |
| ind_5 | FEMALE | ind_3 | ind_2 |
The trait description file contains all information necessary to describe the traits under investigation. Like the experiment description file, the trait description file contains two columns with tag-value pairs. More than one trait can be described by separating them with a blank line.
Data provided in this file (for each trait):
Typical issues with this file:
| id | ISU1 |
| name | fat depth |
| description | thickness of the fat in the back of the animal |
| ontology | PATO|acc0000001 |
| ontology | Evo|acc999999 |
| unit | centimeter |
| id | BW |
| name | birth weight |
| description | weight at birth |
| unit | grams |
| id | carcass_weight |
| name | carcass weight |
| description | weight of the animal after slaughter |
| unit | grams |
Three traits are listed in this example. It is made clear that the id for each of them can be any string and has no other meaning than to be a unique identifier that can be reference in the other MIQAS_TAB files, in particular the phenotypes and results files.
The marker description file contains all information necessary to uniquely identify the genetic markers used in the study. Like the previous two files, this file has two columns. Information for different markers is separated by an empty line.
Data provided in this file:
Typical issues with this file:
| id | CGA | |
| name | CGA | |
| accession | ArkdB|ARKMKR00001 | |
| allele | 0 | unknown |
| allele | 1 | 137 bp |
| allele | 2 | 188 bp |
| allele | 3 | deletion |
| id | S0082 | |
| accession | dbSNP|rs0002 | |
| accession | ArkDB|ARKMKR00002 | |
| allele | 0 | unknown |
| allele | 1 | A |
| allele | 2 | G |
| id | S0083 | |
| accession | dbSNP|rs00003 | |
| allele | 0 | unknown |
| allele | A | A |
| allele | G | G |
| id | S0085 | |
| accession | dbSNP|rs00004 | |
| id | SW1828 | |
| accession | dbSNP|rs00005 |
In this example, the first marker will be referred to in the other files as CGA, i.e. the value for the id field. Four different alleles are described. The values 0 to 3 can be used in the genotype data file and refer to the actual alleles as provided in the third column. These values can e.g. be fragment lengths if the marker is a microsatellite. The second marker is a SNP and has three alleles. This marker has been defined in two different databases that each contain information on it. The third example marker shows that anything can be put in the second column of the allele fields: even the actual biological information. This means that the anonymous label “A” in the genotype.txt file for this marker actually means adenosine. Finally, the last two markers do not list any alleles. In this case, the alleles in the genotypes.txt file must be considered as just anonymous labels.
The genotype data file contains the genotypes for all markers for all individuals. The first column contains the individual id (referring to the individual id in the pedigree file) and the other columns contain genotypes for the different markers. The first row is a header row and should consist of ”#individual_id” in the first cell, followed by marker ids. These marker ids should correspond to the id field in the marker description file; if the marker id cannot be found back in that file, the genotype file is invalid. The values for the genotype recordings must be present in the second column of the “allele” fields in the marker description file. The same marker id should appear once for each allele; for diploid organisms, there should therefore be two columns for each marker.
Typical issues with this file:
| #individual_id | CGA | CGA | S0082 | S0082 | S0155 | S0155 | SW1301 | SW1301 |
| ind_1 | 1 | 4 | 4 | 7 | 1 | 2 | 1 | 2 |
| ind_2 | 1 | 4 | 4 | 3 | 1 | 1 | 1 | 2 |
| ind_3 | 0 | 0 | 0 | 0 | 1 | 3 | 1 | 4 |
| ind_4 | 1 | 2 | 2 | 2 | 1 | 1 | 2 | 2 |
| ind_5 | 1 | 1 | 1 | 2 | 3 | 3 | 2 | 3 |
The phenotype data file is similar to the genotype data file and contains the values for phenotype recordings of the individuals of the study. This file is also grid-based. The first column contains the individual id (referring to the individual id in the pedigree file) and the other columns contain values for the different traits. The first row is a header row and should consist of ”#individual_id” in the first cell, followed by trait ids. These trait ids should correspond to the id field in the trait description file; if the trait id cannot be found back in the trait description file, the phenotype file is invalid. The values for the phenotype recording must be in the unit as recorded in the trait description file.
Typical issues with this file:
| #individual_id | BW | carcass_weight | ISU1 |
| ind_1 | 67800 | 382100 | 19 |
| ind_2 | 75400 | 283100 | 24 |
| ind_3 | 69800 | 235500 | 29 |
| ind_4 | 64800 | 318260 | 18 |
| ind_5 | 65800 | 291700 | 19 |
If the MIQAS experiment describes a QTL study, the genetic map has to be provided in the map file. This file, in contrast to the previous three files, contains a grid with the first column containing the marker identifiers (i.e. the value for id in the marker description file), the second column contains the linkage group and the third column contains the position in centiMorgan. The first row contains the headers: ”#marker_id”, “linkage_group” and “position”. For each linkage group, there should be a marker at zero centiMorgan.
Typical issues with this file:
| #marker_id | linkage_group | position |
| S0083 | 1 | 0 |
| CGA | 1 | 42 |
| S0082 | 1 | 70 |
| S0085 | 7 | 0 |
| SW1828 | 7 | 106 |
A results file contains all QTLs or associated markers that are identified within a single analysis. As with other tag-value files described above, different results (i.e. QTLs or associated markers) are separated by an empty line. The first block of rows describes the analysis and ends with a blank line. Subsequent lines report QTLs or associations, each separated with an empty line.
Data provided in this file:
Typical issues with this file:
| id | results_a |
| trait | BW |
| trait | carcass_weight |
| genetic_model | additive |
| software | GridQTL |
| approach | least square regression interval mapping |
| assumption | effect same in males as in females |
| fixed effect | sex |
| trait covariate | ISU1 |
| qtl id | qtl_1 |
| trait | BW |
| linkage group | 1 |
| region start | 7.2 |
| region stop | 9.3 |
| region peak | 8.1 |
| confidence interval start | 5.2 |
| confidence interval stop | 13.1 |
| confidence interval method | bootstrapping |
| statistical significance | P<0.05 |
| proportion explained | 19.1% |
| significance term | genome-wide significant |
| significance criterion | LOD > 3 |
| qtl id | qtl_2 |
| trait | BW |
| linkage group | 1 |
| region start | 19.2 |
| region stop | 23.3 |
| region peak | 19.9 |
| confidence interval start | 15 |
| confidence interval stop | 28 |
| confidence interval method | bootstrapping |
| statistical significance | P<0.05 |
| proportion explained | 5.4% |
| significance term | chromosome-wide suggestive |
| significance criterion | LOD > 3 |
| id | results_b |
| trait | BW |
| trait | carcass_weight |
| genetic model | additive |
| software | GridQTL |
| approach | least square regression interval mapping |
| fixed effect | sex |
| trait covariate | ISU1 |
| qtl id | qtl_3 |
| trait | BW |
| linkage group | 1 |
| region start | 5.4 |
| region stop | 11.2 |
| region peak | 8.6 |
| confidence interval start | 3.1 |
| confidence interval stop | 14.2 |
| confidence interval method | bootstrapping |
| statistical significance | P<0.05 |
| proportion explained | 14.1% |
| significance term | genome-wide significant |
| significance criterion | LOD > 3 |
Three QTLs are described in this example, separated into two files. The analysis for both files is exactly the same, except that the first analysis (in results_a) uses an assumption that is not present in the second analysis. Therefore, these must be separated into different files. The analysis part of these files should contain all traits that are included in the analysis, even if no QTLs or associated markers could be identified for them. Assumptions should be very short (e.g. “effect same for male and female”) and not contain multiple assumptions in one row. Multiple assumptions should be split over multiple rows.
Values for fixed effects and trait covariates are either (a) traits that are described in the trait description file (and linked through the id field), or (b) any header in the pedigree file (i.e. sex, father_id or mother_id). Values for QTL covariates have to be QTL IDs as specified in the trait description file.
Figure 1: Overview of experiment design that is covered by the MIQAS_TAB specification.